Aujourd’hui bien étudiées et identifiées, les neuropathies périphériques sont devenues un domaine majeur de la neurologie moderne. Toutefois, la grande variété des causes et des formes rend les diagnostics et les traitements encore difficiles. L’AFNP a fait le choix de s’intéresser en priorité aux neuropathies périphériques dysimmunitaires acquises, aiguës et chroniques.
PIDC
Les Polyradiculonévrites Inflammatoires Démyélinisantes Chroniques (PIDC) ou Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP) ou Polyradiculonévrites chroniques (PRNc) constituent une entité qui existe depuis moins de trente ans. Différentes des formes aigües (SGB, AMSAN,AMAN) et des neuropathies associées aux dysglobulinémies, leur origine dysimmune est très probable.
Un peu d’histoire
Guillain découvreur du SGB avait dès 1953 observé des formes chroniques de neuropathies démyélinisantes. Ce n’est qu’en 1975 que Dick et ses collaborateurs décidèrent de regrouper sous une même entité les CIDP différenciées du syndrome de Guillain-Barré. Une absence d’infection récente, avec des signes cliniques évoluant au-delà de 6 mois et présentant une sensibilité aux corticoïdes ont progressivement permis d’élaborer une classification homogène de ces maladies. Les mécanismes inflammatoires sont probablement d’origine dysimmunitaire, mais c’est encore mal connu. La forme commune des PRNc est dite idiopathique (sans cause connue).
Deux formes sont aujourd’hui identifiées: A rechutes et progressives.
Signes et évolution
Si dans 80% des cas , le déficit moteur prédomine, le processus de démyélinisation est aléatoire et multifocal et des asymétries sont observées. Ainsi, le Syndrome de Lewis-Sumner présente des blocs de conduction multifocaux et persitants conduisant à s’interroger à son rattachement identitaire (PIDC? NMM?). Les PIDC peuvent présenter différentes formes. Sensitives, multifocales (SLS), avec signes centraux, avec formes axonales, avec gammapathie monoclonale.
Traitement
Trois traitements ont pu faire la preuve de leur efficacité :les corticostéroïdes, les échanges (EP) plasmatiques et les immunoglobulines intraveineuses (Ig-IV). Bien que leur efficacité ne soit pas réellement démontrée, on peut associer en seconde intention des traitements immunosuppresseurs.
Dans tous les cas, la qualité de vie du malade devra prévaloir au choix thérapeutique qui devra être proposé par le médecin-spécialiste, en partenariat avec le malade.
Quelle attitude thérapeutique?
Il n’existe pas aujourd’hui de critères certains pour prévoir l’efficacité des trois traitements. De nombreux formes particulières de PIDC ont été identifiées en quelques années et il faudra affiner leur caractérisation afin de proposer la meilleure thérapeutique possible.
Syndrome de Lewis-Sumner
C’est en 1982 qu’un sous-groupe de neuropathies sensitivo-motrices chroniques présentant des blocs de conduction persistants a été individualisé.
Le syndrome de Lewis et Sumner(SLS) est une neuropathie démyélinisante, multifocale, sensitivo-motrice, d’origine acquise et dysimmunitaire. Sa fréquence est environ 5 fois moins importante que les PIDC communes et représente environ 1 à 4 cas par million d’habitants. Il se caractérise par une atteinte sensitivo-motrice asymétrique sur la partie distale du membre supérieur, le plus souvent dans le territoire médian et/ou cubital. L’ENMG révèle des anomalies de conduction très focalisées sous forme d’un nombre élevé de blocs de conduction moteurs persistants. Le contexte clinique le plus fréquent est l’apparition d’une amyotrophie associée à un syndrome douloureux et siégeant sur l’un des deux membres supérieurs. Une atteinte des membres inférieurs est possible sous forme de steppage et de paresthésie distale asymétrique.
Le SLS est souvent sensible aux immunoglobulines intraveineuses (IgIV) et sous-cutanées. Les échanges plasmatiques et la corticothérapie ne sont aujourd’hui pas des traitements recommandés. Plusieurs études ont même suggérées un risque d’aggravation sous corticoïdes.
Le traitement par voie intraveineuse peut se faire en milieu hospitalier ou à domicile à condition que la surveillance biologique et clinique du malade soit assurée (collaboration spécialiste, malade, prestataire de service, hôpital…). Le traitement par voie sous-cutanée plus récent offre davantage d’autonomie au malade.
 SLS et NMM ont en commun une atteinte multi focale et la présence de blocs de conduction persistants.
SLS et NMM ont en commun une atteinte multi focale et la présence de blocs de conduction persistants.
Rencontre exceptionnelle entre l’AFNP et les Dr Lewis et Sumner lors du congrès de la Peripheral Nerve Society (PNS) qui s’est tenu à Saint-Malo en juillet 2013 .
Syndrome de Guillain-Barré
De la famille des polyradiculonévrites aigües, il est la cause la plus fréquente des paralysies aigües par atteinte neuromusculaire dans les pays occidentaux. Si les causes exactes de la pathologie ne sont pas connues, des circonstances favorisantes ont été identifiées. 2/3 des malades ont en effet soufferts d’une maladie inféctieuse aigüe, le plus souvent du système respiratoire ou d’une gastro-entérite dans les jours ou semaines qui précède le début de la maladie.
Signes cliniques
Le SGB débute typiquement par des paresthésies des doigts et des orteils, puis une faiblesse des membres inférieurs et une atteinte motrice inconstante des membres supérieurs, de la face et de l’oropharynx.
L’évolution et la sévérité de la neuropathie sont variables. L’installation des paralysies peut être très rapide, au point de nécessiter une intubation et une assistance ventilatoire 24 h à 48 h après l’apparition des premiers symptômes.
Une origine dysimmunitaire
Le SGB résulte d’une anomalie des réponses immunitaires. La gaine de myéline est la cible première de ce dysfonctionnement, dans de rares cas, cela peut être l’axone. Toutefois, le SGB est une maladie auto-immune réactionnelle régressant spontanément. Lorsque les réactions immunes s’arrêtent, la régénérescence et la remyélinisation débutent et aboutissent à une régression rapide et quasi complète des paralysies.
Prise en charge
Hospitalisation toujours dans une unité de soins intensifs. 25 à 30% des malades auront besoin d’une assistance respiratoire. Dès que possible rééducation fonctionnelle. Les échanges plasmatiques et les perfusions d’immunoglobulines à très fortes doses sont aujourd’hui les deux types d’immunothérapie utilisée dans le SGB. Les corticïdes ne sont aujourd’hui pas considérés comme un traitement majeur du SGB.
Évolution
Dans le cas de certaines formes sévères, la durée d’évolution peut être plus longue, du fait de la destruction et de la lenteur de régénérescence axonale. A un an près de 70% des patients ont complétement récupéré. 25% gardent un handicap. Le SGB tue encore dans 5 à 8% des cas. La fatigue est un élément fondamental à prendre en compte, en effet environ 80% des patients récupérant d’un SGB en souffriront durablement.
Neuropathies Motrices Multifocales
Sous groupe de neuropathies sensitivo-motrices les NMM présentent des blocs de conduction persistants qui sont localisés essentiellement sur les nerfs moteurs.
Signes cliniques
Le membre supérieur est presque toujours touché (en particulier nerf radial, médian et cubital). Un déficit moteur marqué sans amyotrophie est évocateur de la maladie au moins dans sa forme précoce. Dans les territoires innervés il peut y avoir des crampes, fasciculations moins souvent des paresthésies et des douleurs . Il n’y a habituellement aucun déficit sensitif. Toutefois, lorsqu’il est présent il est généralement au second plan derrière les déficit moteurs. Des signes sensitifs plus évidents doivent orienter vers le diagnostic d’un Syndrome de Lewis-Sumner.
Une maladie probablement dysimmunitaire
Les mécanismes de la maladie sont méconnus mais la réponse aux traitements immunosuppresseurs et la présence d’une famille d’anticorps (antigangliosides) permettent d’envisager une origine dysimmunitaire.
Evolution
Habituellement lentes et progressives, les NMM peuvent parfois évoluer par poussées suivies de phases de rémission.
Traitements
Les immunoglobulines intraveineuses -IG-IV- ont fait la preuve de leur efficacité. Elles peuvent être associées à des immunosuppresseurs. L’attitude thérapeutique élaborée dépendra essentiellement de la qualité de la réponse aux IG-IV.
Syndrome de Miller-Fischer
Décrit pour la première fois en 1956 par Miller Fischer, ce syndrome est une variante du Syndrome de Guillain-Barré associant typiquement des troubles de la sensibilité (paresthésies), des anomalies dans la coordination des mouvements (ataxie) et des déficits moteurs habituellement observés au niveau des nerfs oculomoteurs avec une vision double ou trouble. Il existe également parfois des troubles de la déglutition dus à une atteinte des nerfs crâniens.
Neuropathies associées aux dysglobulinémies
Polyneuropathie à anticorps anti-Myelin Associated Glycoprotein (MAG)
La polyneuropathie à anticorps anti-MAG (PNP anti-MAG) est une neuropathie dysimmune démyélinisante chronique.
Physiopathologie
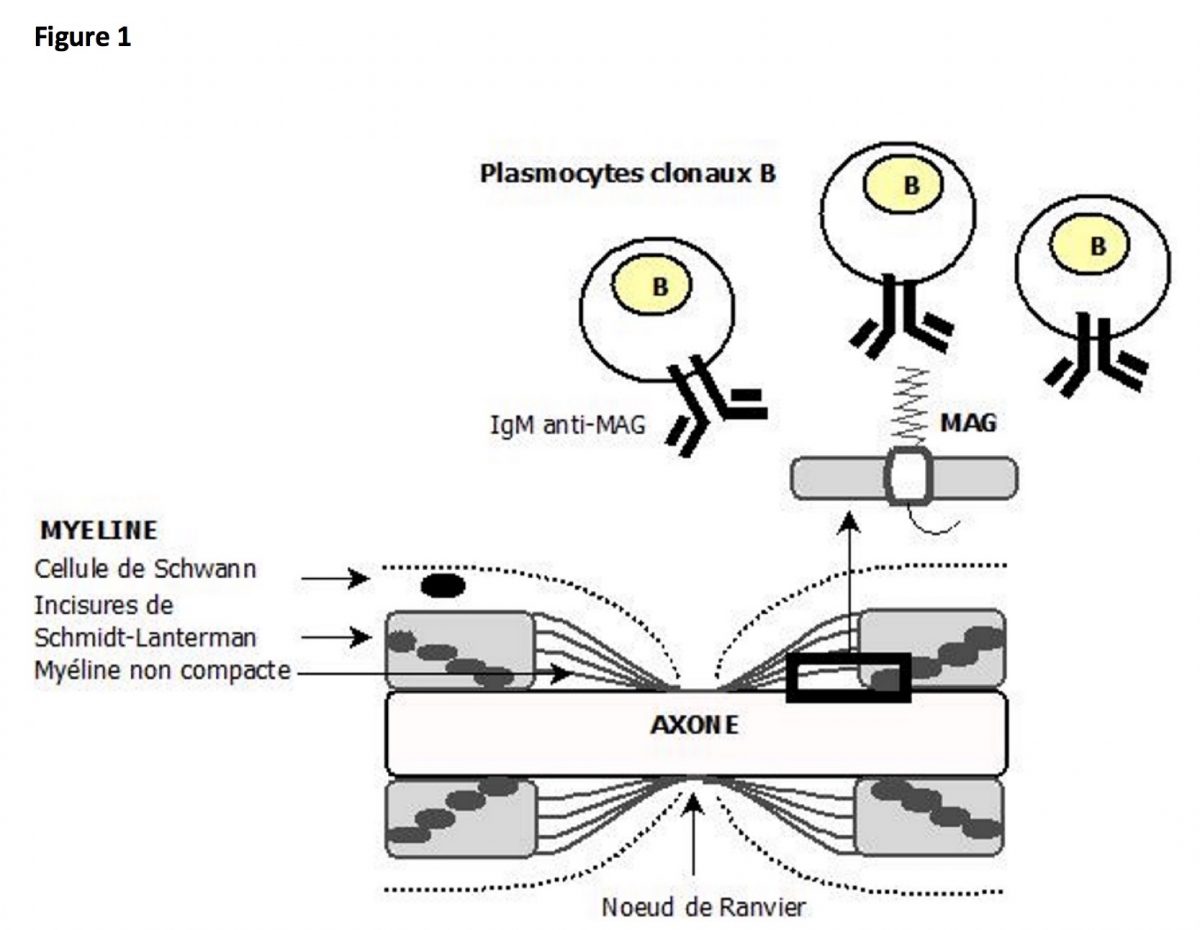
La MAG est une glycoprotéine membranaire située au sein de la myéline non compacte des paranoeuds et des incisures de Schmidt-Lanterman (figure 1). Elle joue un rôle dans la compaction de la myéline, dans l’adhésion entre l’axone et la cellule de Schwann et dans le contrôle du diamètre axonal. Les anticorps anti-MAG sont des immunglobulines d’isotype IgM dirigées contre la MAG. Ils sont produits par une population clonale de plasmocytes B (figure 1), soit dans le cadre d’une gammapathie monoclonale dite bénigne (MGUS), soit dans le cadre d’une maladie de Waldenström.
Clinique
Il s’agit d’une neuropathie rare. Elle touche plus fréquemment les hommes (sex ratio d’environ 2-3/1). Elle débute souvent tardivement dans la 6 ème décennie. Classiquement, la polyneuropathie est d’évolution lente et progressive, avec une prédominance de symptômes sensitifs (engourdissement, fourmillements des extrémités, trouble de l’équilibre ou de la coordination= ataxie). Parfois, il s’associe un tremblement d’attitude et d’action. Un déficit moteur peut apparaitre au cours de l’évolution (déficit des releveurs des pieds, voir faiblesse des muscles des mains). Environ 25% des patients présentent une incapacité fonctionnelle significative après 10 ans d’évolution.
Diagnostic
Le diagnostic est réalisé à l’aide des arguments cliniques, biologique (dosage des anticorps anti-MAG dans le sang par une technique ELISA) et électroneuromyographique (EMG). L’EMG montre une neuropathie démyélinisant traduisant le dysfonctionnent de la conduction nerveuse lié à l’altération de la gaine de myéline. La perte axonale est souvent marquée. Rarement, une biopsie de nerf peut être utile au diagnostic. Elle montre des altérations spécifiques (démyélinisation avec aspect de décompaction des lamelles externe de la myéline et dépôts d’IgM sur la gaine de myéline).
Prise en charge thérapeutique
De nombreux traitements immunomodulateurs ou immunosuppresseurs ont été testés dans des essais thérapeutiques dans le but de diminuer la quantité et/ou l’activité des anticorps anti-MAG et/ou des lymphoplasmocytes producteurs. Les résultats sont assez décevants…
Les immunoglobulines ont montré sur le court terme une différence par rapport au placebo mais le bénéfice clinique apparait limité. Le rituximab étudié dans 2 essais (anticorps monoclonal anti-CD20 qui cible les lymphocytes B) a montré chez certains patients une amélioration de plusieurs critères de jugement secondaires comparé au placebo mais il existe des limitations méthodologiques. Lorsque la maladie de Waldenström est symptomatique, une chimiothérapie est proposée au patient par les hématologues. Des polymères « leurres » ressemblant à la MAG et permettant une liaison aux AC anti-MAG présents dans le sang sont en cours de développement.
La prise en charge symptomatique comprend de la kinésithérapie et le traitement d’éventuelles douleurs neuropathiques.
Dr Juliette Svahn-CHU Lyon.
 CANOMAD
CANOMAD
Le syndrome CANOMAD est l’acronyme de Chronic Ataxic Neuropathy with Ophtalmoplegia, M-protein Agglutination and Disialosyl antibodies, et comporte une PNP sensitive ataxiante chronique avec des troubles oculomoteurs. Il est associé à la mise en évidence d’anticorps anti-gangliosides de type IgM surtout de type kappa, qui réagissent avec un épitope se trouvant sur les gangliosides GD2, GD3, GD1b, GT1b, GT1a et GQ1b. On note parfois une activité d’agglutinines froides. Le tableau clinique est évocateur d’un syndrome de Miller Fisher (ophtalmoparésie, aréflexie et ataxie), mais d’évolution chronique, les caractéristiques de la PNP sont celles compliquant une dysglobulinémique IgM à laquelle se surajoutent des épisodes intercurrents d’atteinte oculomotrice pouvant à tort faire évoquer une atteinte du tronc cérébral. Corticothérapie, cyclophosphamide, IVIg, chlorambucil et échanges plasmatiques ont été proposés avec des résultats variables. Le rituximab reste à évaluer dans cette indication.
Le système nerveux périphérique
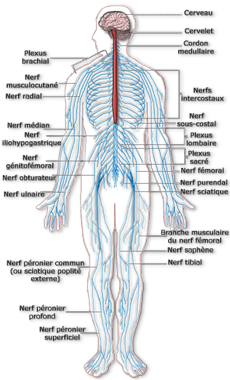
Le système nerveux périphérique (SNP) est un vaste réseau de nerfs qui permet de relier le système nerveux central au reste du corps et de véhiculer des informations sensitives et motrices.
Composition:
– 31 paires de nerfs rachidiens ou spinaux qui naissent dans la moelle épinière et se poursuivent jusque dans les membres.
– 12 paires de nerfs crâniens qui naissent dans les noyaux du tronc cérébral.